- 在多百万次碰撞横截面(CCS)中,在几种与捕获的离子迁移光谱(TIMS)和Pastulation-Seriallial(Pasef)上的捕获离子迁移光谱(TIMS)和Pastulation-Serial-Seriachation(Pasef)的全部蛋白酶横截面(CCS)。
- Large-scale CCS data from 360 LC-TIMS-MS/MS runs, processed with MaxQuant
- 对于CCS对准,跨越347,885肽CCS值,重复测量,中值变异系数(CV)为0.4%;突出显示出在较长时间内和跨仪器的速度CCS的优异再现性
- Precision (CV < 1%) of CCS data is sufficient to train a deep recurrent neural network that accurately predicts CCS values solely based on proteogenomic peptide sequences (R > 0.99)
- 利用深度学习,现在可以预测任何肽和生物的CCS值,形成高级4D蛋白质组学的基础,用于充分利用附加肽CCS信息
MUNICH--(BUSINESS WIRE)--Bruker Corporation(纳斯达克:BRKR)今天宣布从学报Matthias Mann和Fabian Theis群体中出版出来的开创性出版物Nature Communicationswith the title ‘Deep learning the collisional cross sections of the peptide universe from a million experimental values’by Florian Meier et al. (doi.org/10.1038/s41467-021-21352-8)1.
此新闻稿具有多媒体。查看此处的完整版本:https://www.businesswire.com/betway捕鱼游戏下载news/home/20210225005647/cn/
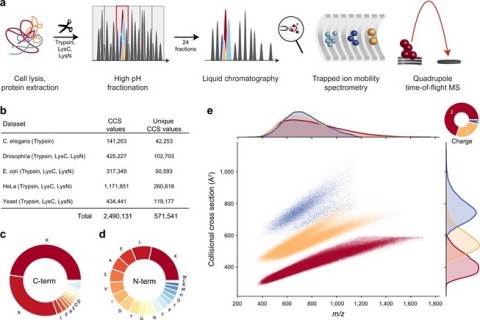
图1:用时和PaseF进行大规模肽碰撞截面(CCS)测量。从“深入了解肽宇宙的碰撞围绕一百万实验值”。(a)通过消化,分馏和每个级分的分馏和色谱分离的整个细胞蛋白质的工作流程。TIMS-四极其TOF质谱仪以PASEF模式操作。(b)通过生物体在本研究中的CCS数据集概述。(c)肽C-末端氨基酸的频率。(d)肽N-末端氨基酸的频率。(e)在CCS与M / Z空间中,在CCS与M / Z空间中的唯一数据点分布为559,979个独特的数据点,包括修改的序列和充电状态。M / Z和CCS的密度分布分别投影在顶部和右轴上。源数据作为源数据文件提供。 (Graphic: Business Wire)
TheNature Communicationspaper describes CCS values measured on the timsTOF pro as an essentially intrinsic property of the peptide ions, which can be used to improve confidence in peptide and protein group identification in 4D shotgun proteomics. Since mass spectrometry-based proteomics relies on accurate matching of acquired spectra against a database of protein sequences, accurate CCS values offer the benefit of narrowing down the list of candidates. This is essential for high sensitivity proteomics where low levels of peptide signals need to be accurately measured in a complex mixture, e.g. in plasma proteomics, peptidomics, immunopeptidomics or metaproteomics.
The publication summarizes a collaborative research effort led by Professor Matthias Mann, who holds dual appointments at the Max Planck Institute of Biochemistry in Martinsried, Germany and the Novo Nordisk Foundation Center for Protein Research at the University of Copenhagen in Denmark, together with the group of Professor Fabian Theis, who also holds dual appointments at the Helmholtz Center Munich in the German Research Center for Environmental Health, and in the Department of Mathematics at TU Munich, in Germany.
Lead author Dr. Florian Meier, now an Assistant Professor in Functional Proteomics at the Jena University Hospital in Germany, said: “The scale and precision of peptide CCS values in our data from the timsTOF pro was sufficient to train our deep learning model to accurately predict CCS values based only on the peptide sequence. This connection between the amino acids contained within a peptide sequence and its measured CCS has tremendous potential to increase the confidence of protein identification. Since the peptide CCS values are entirely determined by their linear amino acid sequences, they should be predictable with high accuracy and our deep learning model accurately predicted CCS values even for previously unobserved peptides. We acquired data from whole-proteome digests of five organisms, which resulted in the measurement of over two million CCS values, including about 500,000 unique peptides, making it by far the most comprehensive CCS data set to date.”
Professor Matthias Mann added: “The source code is publicly available so that further developments can be accelerated for training and prediction models of the human peptide universe. Conceptually, our CCS model could make dia-PASEF faster and less expensive by reducing the effort to generate libraries. Additionally, predicted CCS values should allow for the use of community libraries, such as the Pan Human library, a repository of over 10,000 human proteins, for targeted proteomics.”
Professor Fabian Theis stated: “Deep learning, in particular the used recurrent neural networks need a lot of samples to be predictive, so I was very happy when Matthias approached me and we jointly were able to predict and interpolate biochemical properties of peptides based only on their sequence. I personally liked the fact that we could thus impute CCS values also for many never before measured peptides."
加里Kruppa博士力量副甲氧苄吲酚ent for Proteomics, commented: “This paper showcases the tremendous potential of accurate CCS values for TIMS-PASEF methods in unbiased, deep 4D proteomics. The proven robustness, higher throughput and ultra-high sensitivity of thetimstof.平台非常适合翻译研究。大规模肽CCS值在大型队列研究中蛋白质鉴定和定量的置信度提供了基本优势。此外,CCS值用于提高识别置信度的益处也适用于其他多组合器TIMSTOF工作流程,例如代谢组科,脂质族学和糖类。这些是我们快速增长的TIMSTOF用户社区的令人兴奋的时期。“
About Bruker Corporation(纳斯达克:BRKR)
布鲁克是使科学家能够做出突破性的发现,并开发提高人类生活质量的新应用。Bruker的高性能科学仪器和高价值分析和诊断解决方案使科学家能够在分子,细胞和微观水平下探索生命和材料。必威手机客户端与我们的客户密切合作,Bruker正在实现创新,提高生产力和客户在生命科学分子和细胞生物学研究中取得的成功,在应用和制药应用中,在显微镜和纳米分析以及工业应用中。必威东盟体育Bruker在临床前成像,临床表情研究,蛋白质组学和多组合,空间和单细胞生物学,功能性结构和凝聚态生物学以及临床微生物学和分子诊断中提供差异化的,高价值的生命科学和诊断系统和解决方案。有关更多信息,请访问:www.fatactor.com..
1Meier, F., Köhler, N.D., Brunner, AD.等等。Deep learning the collisional cross sections of the peptide universe from a million experimental values.Nat Commun12,1185(2021)。https://doi.org/10.1038/s41467-021-21352-8

View source version onbusinesswire.com:https://www.businesswire.com/betway捕鱼游戏下载news/home/20210225005647/cn/
媒体:
Petra Scheffer
Bruker Daltonics Marketing & Communications
T:+49(421)2205-2843
E:[电子邮件受保护]
投资者:
Miroslava Minkova
Director, Investor Relations & Corp. Development
Bruker Corporation
T:+1(978)663-3660,EXT。1479.
E:[电子邮件受保护]
Source: Bruker Corporation